Afni: Preprocessing Functional Images
|
|
|
Functional
images are contained in p-files. They are low resolution (64x64)
and need to be: 8) scripts for preprocessing Although all the Preprocessing Steps are the same for Block and Event-Related designs, the examples on this web page assume you are working with the Block Design tutorial files. You may, of course, adapt this guide to other datasets if you prefer. This tutorial assumes that you have afni and grecons5x installed on your sgi or sun machine, and the MGH software (we use fmri.tar). To run the MGH software, you'll need Matlab. You will also need programs that can reconstruct your raw fmri files and rename them. We use grecons5x for reconstructing the files (fMRI P-files that come off the GE 1.5 Tesla Signa 5.x wholebody echospeed Horizon scanner)and a couple of scripts for doing the renaming. See also Scanner Updates for information about preprocessing data gathered after September, 2002. To translate files from Afni format to MGH format, you'll need the afnireg2bshort script. If you need these, download afnireg2bshort,and from the Misc area, download grecons5x (source, irix or linux binary), rename_anat and rename_spiral (unix shell scripts). If you need to rename more than 10,000 files, check out rename_spiral2 in the Misc area. Put the files somewhere reasonable on your machine. Preprocessing scripts to automate it all. I have recently written shell scripts to automate the preprocessing stream. For more info, click here. Starting the Tutorial Make sure you have a fresh directory to work in. Put the Study1Orig.tar.gz into this new area. Cd to this directory to unzip and untar copies of the files from the website: >gunzip Study1Orig.tar.gz >tar xvf Study1Orig.tar Make a subdirectory with an appropriate name (e.g. Here we use the name of the condition during which the functional data was collected): >cd Study1 Note: If you don't have the appropriate grecons5x, you might want to start with the Study1prepped.tar.gz. This data has been greconed and renamed already, so you can start using it with afni right away. Reconstruct Run grecons5x as shown below (note that grecons5x is not available on linux). This will create Slices (17) x # of REPS (# of time points; 80 in this case) number of files (1360 in this case). The command below also removes troublesome B0 (that's B-zero) files. This version of grecon will work correctly for the tutorial set, and most datasets. If you encounter problems (because you have an older dataset), you may need to find an earlier version of grecon. Note that you must have grecons5sx installed for this step to work. greconss5x is not available for linux. >grecons5x
P15872; rm B0* After you have reconstructed the images, you should expect to have thousands of files in the directory, names like so: P15872.001, P.15872.002 etc. Data Collected after September 2002? Your P-files will be named with a .7 at the end which you should treat as part of the name. Expect the output of grecons to be something like this P12345.7.001 etc. See the Updates page. Note that the initial processing of your data (grecons and rename) will be different depending on how big the data set is (see above) and when it was collected. You will need to spend some time checking parameters and making sure you have and are using the correct files. Prep Scripts: In the tools area on Merlin, you will also find versions of prep and prepb for the in_out sequence (prepio, prepbio) and for version 2 of grecons (prep_v2, prepb_v2, prepio_v2, prepbio_v2). To learn more about versions of grecons, see the grecons table. Rename >rename_spiral [enter Pfile name when prompted, e.g., P15872] You should now expect the filenemes to be padded with more zeros. For example, if your original filename was P12345.001, the renamed one might be P12345.0001. All this padding with zeros should make the operating system list the files in the correct order. This is what you want. If you don't rename, then when you construct an image brik, the images will be hopelessly out of order (a very bad thing). Note that all procedures that follow are run from the parent directory so that the resulting files will end up in the parent directory rather than with all the reconstructed P-files. More than 9,999 images? Use rename_spiralb instead. Unfortunately, as of 5/12/2003 this more than 9,999 problem is not resolved for all scripts in the afni processing stream...stay posted. To download the rename_spiralb script, go here: rename_spiralb. See also the Updates page on the More than 9,999 problem. Data Collected after September 2002? Your P-files will be named with a .7 at the end which you should treat as part of the name. Expect the output of grecons to be something like this P12345.7.001 etc. See the Updates page. Note that the initial processing of your data (grecons and rename) will be different depending on how big the data set is (see above) and when it was collected. You will need to spend some time checking parameters and making sure you have and are using the correct files. Prep Scripts: In the tools area on Merlin, you will also find versions of prep and prepb for the in_out sequence (prepio, prepbio) and for version 2 of grecons (prep_v2, prepb_v2, prepio_v2, prepbio_v2). To learn more about versions of grecons, see the grecons table. Make a Functional Brick Go up to the parent directory: >cd .. In the to3d command below: After -time: tz enter the # of reps, # of slices and TR, in that order. To learn more about to3d options, simply type >to3d -help | less at the prompt. In the command below, you call to3d, then tell it you are working with an epan (echoplanar) type file, -prefix is a flag followed by the prefix you want afni to use on the output BRIK, -time is followed by ordering information t=time, z=physical slices, 80=ntr, #timepoints, 17=# of physical slices, 2000=TR, seqplus=sequential in the plus direction. Finally, list the path to the files to be used in the BRIK. >to3d -epan -prefix study1 -time:tz 80 17 2000 seqplus 'Study1/P15872.*' If to3d will not run, you can add the following line to your .cshrc: setenv AFNI_TO3D_OUTLIERS="No" Alternatively, simply include the following flag in your to3d command -skip_outliers However, this does not deal with the real problem, explained below: Afni
will not generate graphs on some unix systems (sgi's are particularly
notorious) because the system reports that it supports the "double
buffer extension" (DBE), but in fact does not. AFNI detects
the support for DBE, but when it tries to use it to draw the outlier
graph, you get the error message. The solution is to set the environment
variable AFNI_NO_XDBE to YES -- see http://afni.nimh.nih.gov/afni/docREADME/README.environment
if you need help on setting such variables. The
relevant section from the README.environtment page is: Once you have this environment variable set, you may want to add the -save_outliers and a filename to your to3d command line: -save_outliers
fname A window will pop up:
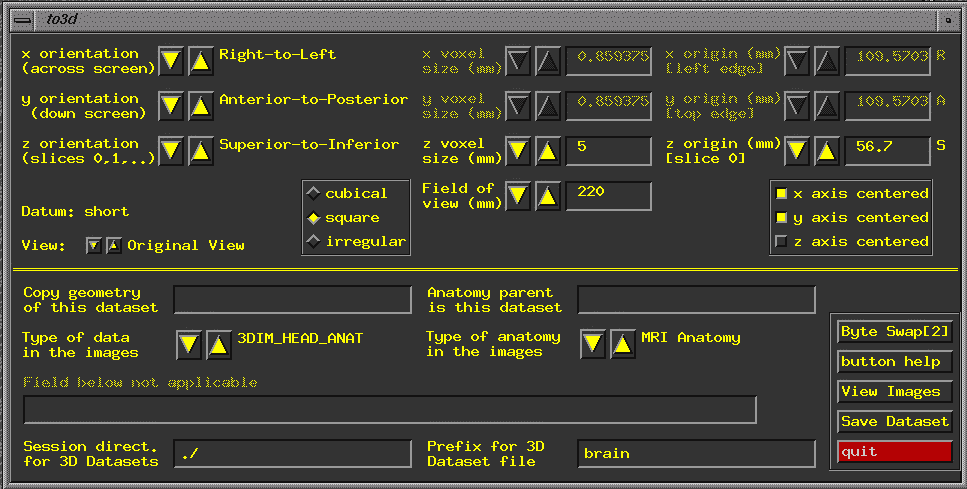
On the lower right is a button "View Images". Press this to see your image. On the upper left, set the orientation of your image. For the tutorial image (which is collected in the standard way for the CNL):
In addition, you need to set voxel dimensions. Our voxels are square in the x-y plane, but the distance between slices is 6 mm, so we choose "square voxels" (the box in the middle of the window), and we set slice thickness (z voxel size) to 6 mm. Our FOV is 220, we set that as well. "Save Dataset", and "quit". Reregister (to correct for minor head movement) >3dvolreg -verbose -Fourier -prefix study1_3dreg -base 3 -dfile study1_3dreg.log study1+orig -verbose
Print progress reports. Use twice for LOTS of output. (Note: This can be done w the gui interface. One can use a different base image (instead of 3) and can even use a run as the basis for other runs. Choose graph "yes" in the gui interface and examine it for moderate flatness to ascertain whether registration has gone pretty well, flat=good) Deconstruct (extract files from AFNI briks prior to MGH formatting) >from3d -input study1_3dreg+orig -prefix study1_3dreg Format (create bshorts from extracted reregistered images; the script afnireg2bshort, aka grecon2bshort is an MGH tool, not part of the afni distribution) >afnireg2bshort -i study1_3dreg -fgs 1 -nas 17 -nfs 80 -i=input, -fgs=first good slice, -nas number of anatomical slices, -nfs number of functional slices (this is the ntr, apparently called the "Number of Temporal Frames" on the scanner console interface) Normalize (note that this must be done on a machine that has matlab, such as Holly, Buddy or Merlin, this is also part of the MGH software): >inorm -i study1_3dreg -o study1_3dreg_norm -i=input, -o=output Create Normalized Brik (this is for the Block Design only) >to3d -epan -prefix study1_norm -time:tz 80 17 2000 seqplus 3D:0:0:64:64:80:study1_3dreg_norm\*.bshort This is like the previous to3d command, except for all the stuff after 3D which specifies the characteristics of the image: 3D:hglobal:himage:nx:ny:nz:fname [16 bit input] hglobal
= number of bytes to skip at start of whole file If you try this command from another directory, you must enclose the directory name in single quotes and get rid of the / before the *.bshort: >to3d -epan -prefix study1_norm -time:tz 80 17 2000 seqplus 3D:0:0:64:64:80:'study1_3dreg_norm *.bshort' Preprocessing Scripts Data Collected after September 2002? Your P-files will be named with a .7 at the end which you should treat as part of the name. Expect the output of grecons to be something like this P12345.7.001 etc. See the Updates page. Note that the initial processing of your data (grecons and rename) will be different depending on how big the data set is (see above) and when it was collected. You will need to spend some time checking parameters and making sure you have and are using the correct files. Prep Scripts: In the tools area on Merlin, you will also find versions of prep and prepb for the in_out sequence (prepio, prepbio) and for version 2 of grecons (prep_v2, prepb_v2, prepio_v2, prepbio_v2). To learn more about versions of grecons, see the grecons table. For more information, see Prep Scripts. Afni Specific Preprocessing Scripts afniprep, afniprepio: These will go through the standard preprocessing steps, from building the first to3d brik, through normalization with inorm and then building a final brik. If you have used a spiralio, then you should use afniprepio. You have the choice to run these with a Swap argument. If you are running on linux, use Swap. otherwise, don't. The commands for running afniprep and afniprepio are the same, but what happens internally is different. You must also provide an argument I call z1: this is the orientation of the z axis, if your images are collected inferior to superior, then z1 should be set to I. If the images are collected Superior to Inferior, then set z1 to S. In the examples below, the images were collected Inferior to Superior. One benefit of these scripts is that you don't have to use the graphical user interface for to3d. The other benefit is simplification, especially if you are running on linux sometimes and sgi at other times. The scripts make some assumptions (like that your images are 64*64, fov=220, images are in radiological orientation and roughly axial, and that you want to run mgh inorm). If you don't like the assumptions, you might still find the scripts useful as a starting point for making something of your own. Usage:
afniprep <Pfile> <nfs> <nas> <output name>
<tr> <thick> <z1> <Swap> afnianat2d and afnianat3d: These scripts will reconstruct 2D and 3D anatomicals respectively. They depend upon the presence of anat_rename3 and afni. They are designed to save you from the to3d gui interface so you can run a bunch of stuff. They make some assumptions about image size 256x256 for 2Ds; 256x256x124 for 3Ds (spgrs) and fov=220 for 2Ds and 250 for 3Ds. Finally, they assume the image names do NOT have the MR at the end (i.e., they are of the form 1234.1.1.). If you are reconstructing on linux, use Swap. You must also provide an argument I call z1: this is the orientation of the z axis, if your images are collected inferior to superior, then z1 should be set to I. If the images are collected Superior to Inferior, then set z1 to S. In the examples below, the images were collected Inferior to Superior. Usage:
afnianat2d <infile> <outfile> <nas> <thick>
<z1> <Swap> Usage:
afnianat3d <infile> <outfile> <Swap> |